
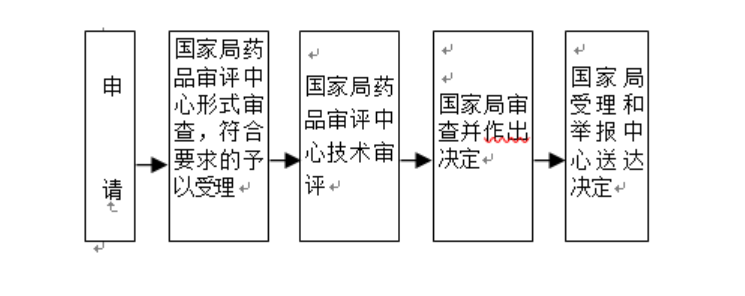
①受理;
②技術審評;
③注冊核查;(審評過程中基于風險啟動)
④注冊檢驗;(審評過程中基于風險啟動)
⑤綜合評價;
⑥行政審批;
⑦行政許可決定;
⑧制證送達。
對于FDA批準的新劑型產品(505b2途徑),且橙皮書顯示該產品為RLD和RS,如申請進口注冊,應按5.1還是5.2類申報?
答: 按照當前法規,此類情形可按5.1類進行申報。
藥品申報資料中臨床試驗報告的封面有哪些要求?
答: 應符合《國家藥品監督管理局關于發布化學藥品注冊受理審查指南(試行)的通告》((2020年第10號)的要求,臨床試驗報告應符合相關指導原則要求。臨床試驗報告標題頁應提供藥品注冊申請人(簽字及蓋章),主要或協調研究負責人(簽字)、復核或協調研究單位名稱、統計學負責人(簽字)和統計單位名稱及ICHE3要求的其他信息;臨床研究報告附錄II中應提供申辦方負責醫學專員簽名。
對于已上市疫苗改變免疫劑量或免疫程序,改變適用人群的應按照什么類型申報?
答: 按照《國家藥監局關于發布生物制品注冊分類及申報資料要求的通告》(2020年第43號)預防用生物制品注冊分類要求,改變免疫劑量和免疫程序屬于注冊分類2.5,改變使用人群屬于注冊分類2.6,應按照藥物臨床試驗和上市許可申請通道進行申報。
申報資料中的外文資料是否全部翻譯為中文?
答: 根據《藥品注冊管理辦法》,全部申報資料應當使用中文并附原文,其他文種的資料可附后作為參考。中文譯文應當與原文一致。
關于按照生物制品管理的體外診斷試劑是否需申報臨床試驗申請?
答: 按照生物制品管理的體外診斷試劑,申請人在境內完成臨床試驗后可以直接提出藥品上市許可申請。
按照生物制品管理的體外診斷試劑申報資料是否按照M4:人用藥物注冊申請通用技術文檔(CTD)整理?
答: 應按照《國家藥監局關于發布生物制品注冊分類及申報資料要求(試行)的通告》(2020年 第43號)第三部分 按生物制品管理的體外診斷試劑申報資料要求整理。
關于境外生產的體外診斷試劑臨床申報資料要求?
答: 至少在3家境內臨床機構完成臨床試驗并提供臨床試驗協議及臨床試驗方案。境外申請人應提供在境外完成的臨床試驗資料、境外臨床使用情況的總結報告和在境內完成的臨床試驗資料。










 客服1
客服1