
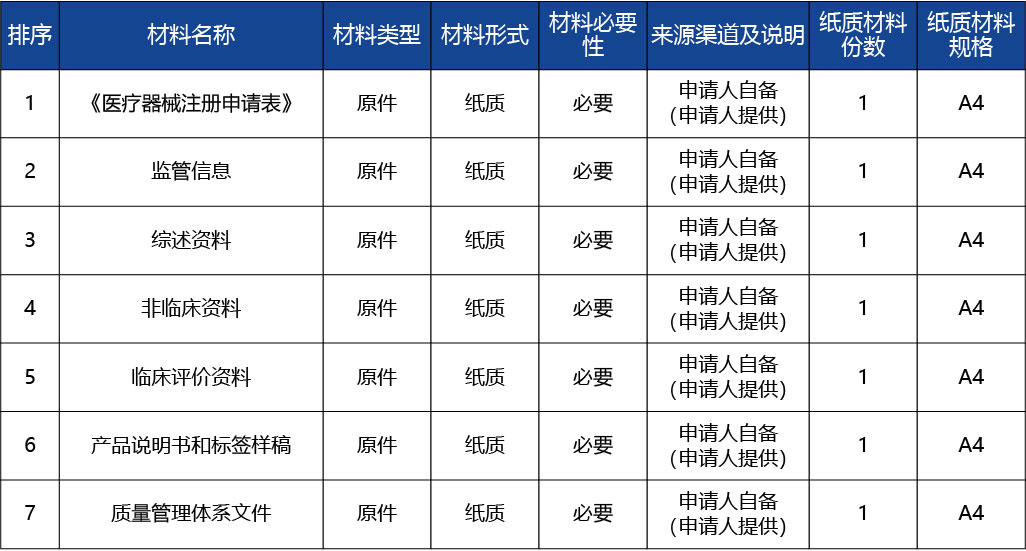
條件名稱:申請事項屬于本部門職權范圍,申報資料齊全、符合形式審查要求。
適用類型:通用
受理1
審查標準:對申請人提交的申請材料不齊全或者不符合形式審查要求的,當場告知申請人補正有關材料。
審查結果:重慶市藥品監督管理局受理通知書
完成時限:0個工作日
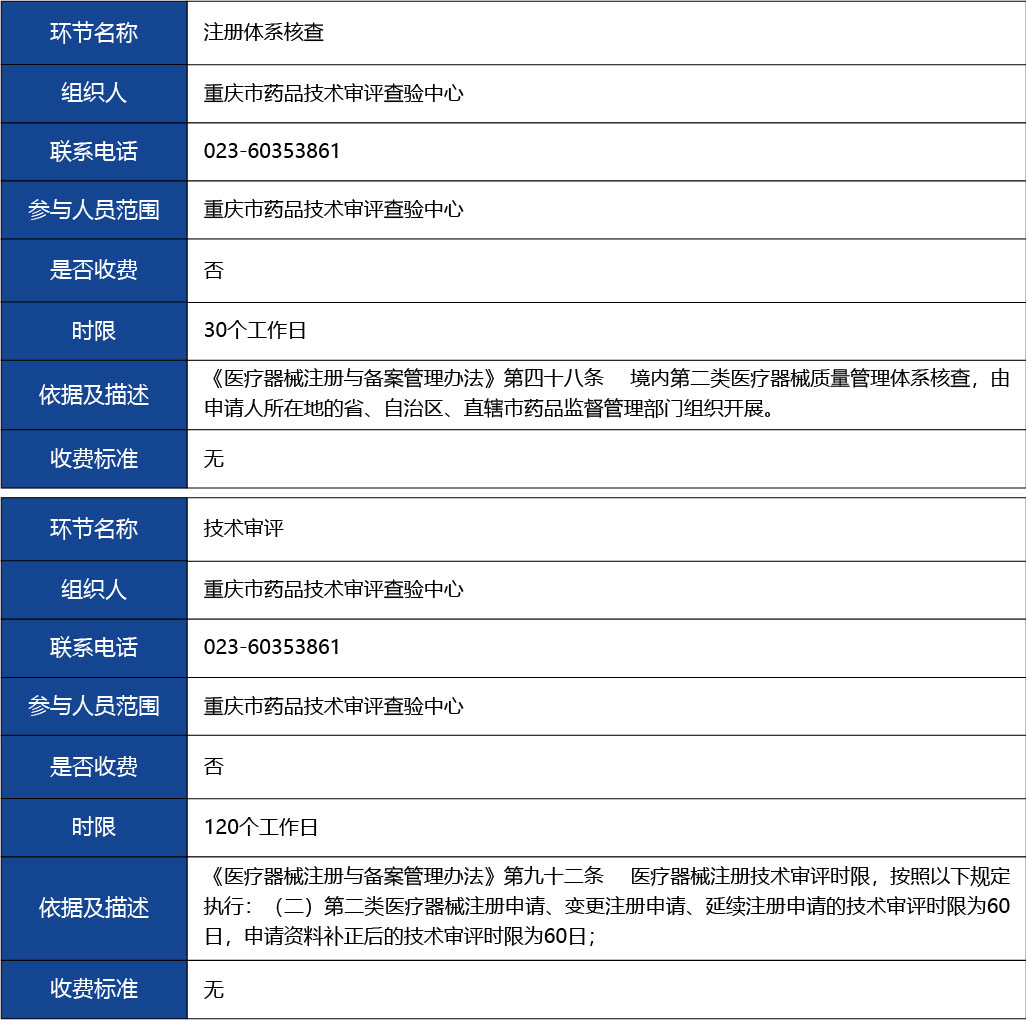
注冊體系核查2
參與人員范圍:重慶市藥品技術審評查驗中心
是否收取費用:否
時限(單位:工作日):30個工作日
依據及描述:無
收費標準:無
技術審評3
參與人員范圍:重慶藥品技術審評認證中心
是否收取費用:否
時限(單位:工作日):120個工作日
依據及描述:無
收費標準:無
審查4
審查標準:對行政審批申請事項的合法性、合理性進行審查,對照審批條件、標準,根據情況分別作出審查意見。
審查結果:醫療器械注冊處產品注冊資料審查意見表
完成時限:5個工作日
決定5
審查標準:對行政審批申請事項的合法性、合理性進行審查,對照審批條件、標準,根據情況分別作出審查決定。
審查結果:醫療器械注冊證審批表
完成時限:5個工作日
送達6
送達方式:申請人自行領取/郵寄
頒發證件:中華人民共和國醫療器械注冊證
辦理期限:0個工作日
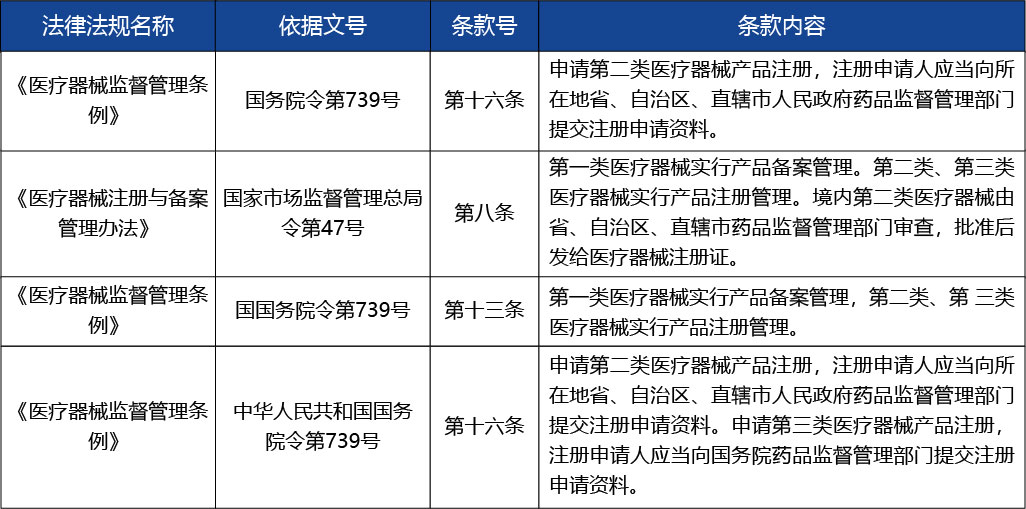

問題:1. 醫療器械注冊過程中主要法律、法規、規章制度?
解答:《醫療器械監督管理條例》、《醫療器械注冊與備案管理辦法》、《醫療器械通用名命名規則》、《醫療器械臨床試驗質量管理規范》、《醫療器械生產質量管理規范》、《醫療器械說明書和標簽管理規定》、《醫療器械注冊申報資料要求和批準證明文件格式的公告》等。
問題:開展醫療器械臨床試驗有哪些規定
解答:開展醫療器械臨床試驗,應當按照醫療器械臨床試驗質量管理規范的要求,在取得資質的臨床試驗機構內進行。臨床試驗樣品的生產應當符合醫療器械質量管理體系的相關要求。第三類醫療器械進行臨床試驗對人體具有較高風險的,應當經CFDA批準。需進行臨床試驗審批的第三類醫療器械目錄由CFDA制定、調整并公布。臨床試驗審批是指CFDA根據申請人的申請,對擬開展臨床試驗的醫療器械的風險程度、臨床試驗方案、臨床受益與風險對比分析報告等進行綜合分析,以決定是否同意開展臨床試驗的過程。醫療器械臨床試驗應當在批準后3年內實施;逾期未實施的,原批準文件自行廢止,仍需進行臨床試驗的,應當重新申請。










 客服1
客服1