NMPA注冊
FDA注冊
CE注冊
海外注冊
(GCP)相關
倫理相關
臨床研究(IIT)
相關原則
預試驗的標準
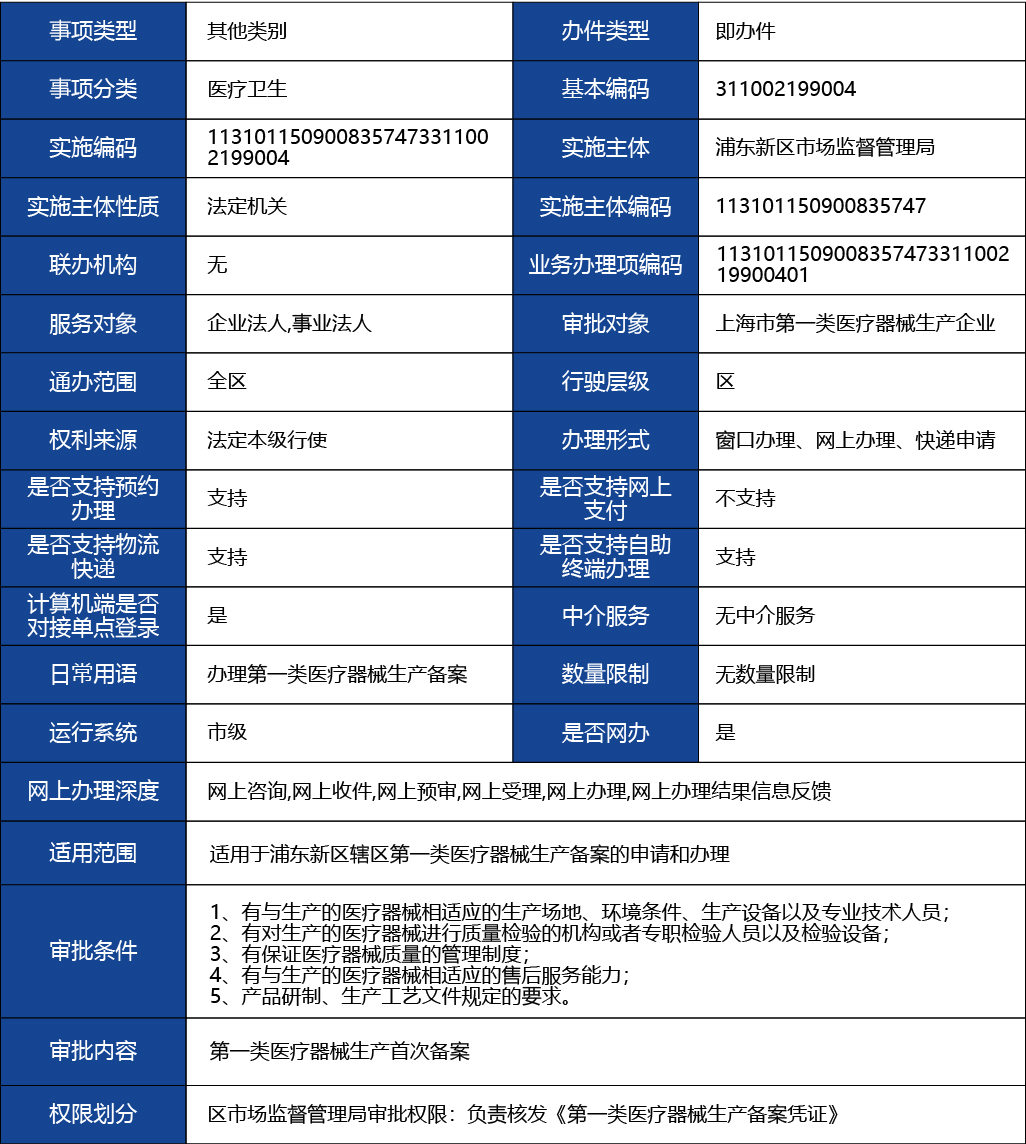
醫療器械注冊
備案資料符合形式審查要求的,當場準予備案。
填報須知
可下載示例樣表查看具體要求。
形式標準
* 如未開展全程網辦,備案人需上傳所有電子備案資料,并通過法人一證通簽章(如企業需要同時上傳Word版本文件,可將蓋章掃描件連同word版本文件打包后上傳,此時無需再對壓縮包文件進行法人一證通簽章)、并遞交所有已加蓋企業公章的紙質備案資料,紙質資料遞交要求如下:1、遞交資料中有復印件的,需在復印資料上加蓋“內容與原件一致”的章。(或可手寫并簽字確認)2、遞交資料需加蓋單位公章(及騎縫章)。3、申報資料按順序排列、裝訂成冊,并制訂資料目錄(有1、2級標題;以表格形式說明每項的卷和頁碼)。* 如已開展全程網辦,備案人只需上傳所有電子備案資料,并通過法人一證通簽章(如企業需要同時上傳Word版本文件,可將蓋章掃描件連同word版本文件打包后上傳,此時無需再對壓縮包文件進行法人一證通簽章)。
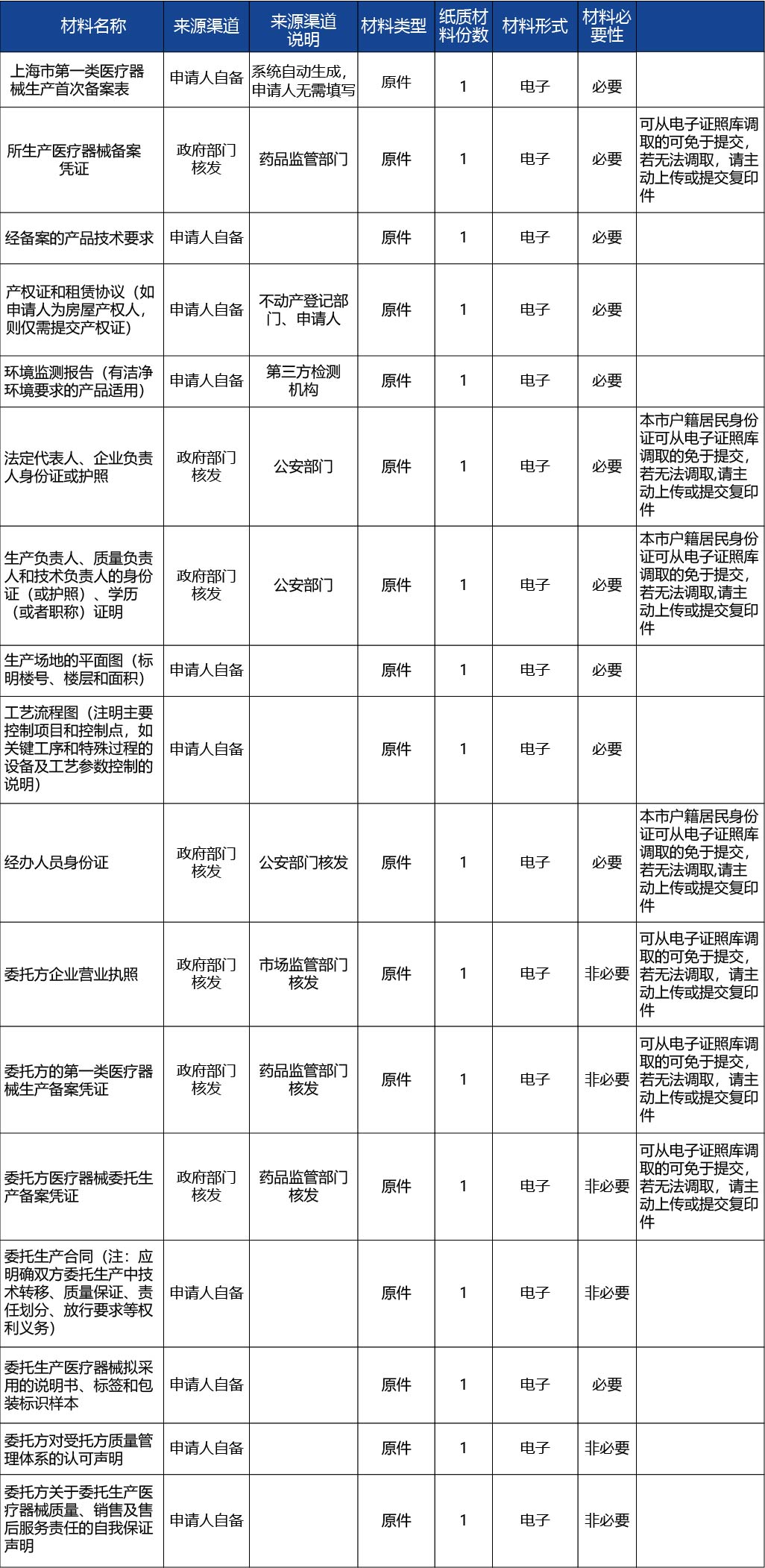
申請材料目錄
我的免交材料
注:本市政府部門核發材料可通過電子證照、數據核驗等方式免提交,具體以實際辦理情況為準。
申請文書名稱
第一類醫療器械備案表(首次備案)
辦理方式
1.收件。申請人通過 “一網通辦”統一受理平臺、物流快遞等方式提交申請材料。收件工作人員依據申請材料目錄及形式標準,對申請人提交的申請材料進行審核。
2.受理。受理工作人員依據受理條件對申請材料進行審查,對申請材料齊全、符合法定形式的,予以受理;對申請材料不齊全或者不符合法定形式的,一次告知申請人需要補正的全部內容。
3.審查 。經辦人員當場對申請材料進行審查。
4.決定。經辦人員對申請材料齊全、符合法定形式的,當場作出準予許可的決定。
5.制證與送達。經辦人員根據審批決定,當場制作第一類醫療器械備案憑證并送達申請人(或根據申請人要求通過物流快遞等方式送達)。
特別程序
不含有特別程序
中華人民共和國國務院令第739號《醫療器械監督管理條例》第十三條 “第一類醫療器械實行產品備案管理,第二類、第三類醫療器械實行產品注冊管理。”中華人民共和國國務院令第739號《醫療器械監督管理條例》第十五條 “第一類醫療器械產品備案,由備案人向所在地設區的市級人民政府負責藥品監督管理的部門提交備案資料。”
是否收費:否
完成備案變更后,獲得何種批件?
對予以備案變更的第一類醫療器械及體外診斷試劑,不再核發備案憑證,但在上海市藥品監督管理局政務網站公示備案變更信息。
完成第一類醫療器械產品備案后,獲得何種批件?
對予以備案的第一類醫療器械及體外診斷試劑,核發《第一類醫療器械備案憑證》,并在上海市藥品監督管理局政務網站公示備案信息。對于已經開展全程網辦的區市場監督管理局,備案人可在申報平臺下載《第一類醫療器械備案憑證》的電子簽章文件。
辦理備案變更時,備案表“變更情況”欄目的內容如何填寫?
應簡明扼要具體表述變更內容,因為這部分信息是對外公示的內容,表述不清晰會給后續的產品銷售、備案后監管帶來困惑。例如:變更情況僅填寫“有效期變更”則過于簡單。可填寫變更后內容,例如有效期變更為×××,或者填寫變更前后比對內容,例如有效期由“×××”變更為“×××”。
何種細胞培養基可以按第一類體外診斷試劑進行產品備案?
僅用于細胞增殖培養,不具備對細胞的選擇、誘導、分化功能,培養后的細胞用于體外診斷的細胞培養基屬于第一類體外診斷試劑。細胞培養基類產品備案,產品名稱應為“XX培養基”,并根據產品實際情況,參照目錄中的培養基產品描述其預期用途,預期用途中應包含“僅用于細胞增殖培養,不具備對細胞的選擇、誘導、分化功能,培養后的細胞用于體外診斷”的內容。
只有列入《體外診斷試劑分類子目錄》的第一類體外診斷試劑才可以進行產品備案么?
除《體外診斷試劑分類子目錄》中列入的屬于第一類體外診斷試劑的染色液類和微生物培養基類產品外,其他所有染色液類產品、不用于微生物鑒別和藥敏鑒別的微生物培養基均屬于第一類體外診斷試劑。該類產品備案,產品名稱應為“XX染色液”或“XX培養基”,并根據產品實際情況,參照目錄中的相關產品描述其預期用途(其中微生物培養基類產品應不具有微生物鑒別和藥敏鑒別的作用)。
屬于第一類醫療器械的組合包類產品,如何進行產品備案?
由需配合使用從而實現某一預期用途的一種以上醫療器械組合而成的產品,若組合中所有產品均為第一類醫療器械(不得含有任何形式的非醫療器械產品),且組合后不改變各組成器械的預期用途,可按照第一類醫療器械備案。其產品名稱應體現組合特性,原則上按其主要臨床預期用途命名,名稱的組成內容應在所屬相關目錄“產品類別(一級或者二級)”、所含各產品的“預期用途”范圍內,如上肢內固定手術器械(包)、膝關節手術器械(包)等。同時,“產品描述”應包含所有組成的醫療器械,并說明各組成醫療器械的“產品描述”和“預期用途”,且其基本內容均應與目錄中的相應內容一致。
如相關產品屬于《醫療器械分類目錄》(2017版)中的第一類醫療器械,辦理產品備案時,產品描述以及預期用途必須與目錄中的產品描述以及預期用途內容完全一致么?
《醫療器械分類目錄》中的內容是對按第一類醫療器械管理的產品的原則要求,備案人應針對備案的產品進行具體描述,不建議使用“通常”、“一般”等比較含糊的詞匯,同時確保不超出目錄中“產品描述”相關內容的范圍;“預期用途”的基本內容應與目錄中的相應內容一致。
如相關產品屬于《醫療器械分類目錄》(2017版)中的第一類醫療器械,辦理產品備案時,產品名稱必須與品名舉例一致么?
根據原國家食品藥品監督管理總局辦公廳下發的《關于實施第一類醫療器械備案有關事項的通知》(食藥監辦械管〔2014〕174號),實施備案的醫療器械,應首先根據其“產品描述”和“預期用途”的實際情況,通過與目錄中“產品描述”和“預期用途”的內容綜合判定產品的歸屬類別,包括所屬子目錄、一級及二級類別。根據所屬類別,應直接使用目錄中“品名舉例”所列舉的名稱。
如何查詢本市已備案的第一類醫療器械產品相關備案信息?
可至上海市藥品監督管理局政務網站(yjj.sh.gov.cn),在政府信息公開----數據查詢----產品查詢----“上海市第一類醫療器械備案數據庫”欄目進行查詢。
對于首次備案、備案變更情形,備案表“符合性聲明”欄目填報內容其中一項為“聲明本產品屬于第一類醫療器械產品,并寫明確切的分類依據”,如何填寫確切的分類依據?
根據引用的不同目錄或文件填報信息,以下為原則性要求:1)2014年國家局下發的《第一類醫療器械目錄》(需注明所屬子目錄以及相應序號)2)《醫療器械分類目錄》(2017版)(需注明所屬子目錄、序號以及二級產品類別)3)《體外診斷試劑分類子目錄》(2013版)(需注明序號以及產品類別)4)《關于過敏源類等體外診斷試劑產品屬性及類別調整的通告》(2017年第226號)(需注明哪一個附件以及相應序號)5) 國家局于2014年6月1日之后下發的分類界定文件(需注明具體的文號、文件名、引用的具體內容)6)“醫療器械分類界定信息系統”平臺中的《分類界定告知書》(需注明受理號、告知號以及《分類界定告知書》中相關產品信息的具體內容)7)如有其他相關依據也要注明具體出處和內容。
常見錯誤示例
1、常見錯誤:資料未按照格式要求填寫,字體大小不符合要求。正確做法:按照申請表要求正確填寫。
第一類醫療器械產品首次備案
《醫療器械監督管理條例》第十三條 第一款第一類醫療器···
備案人應當加強醫療器械全生命周期質量管理,對研制、···
《醫療器械監督管理條例》第十三條 第一類醫療器械實行···
天之恒依托醫療器械注冊認證、臨床研究經驗豐富的全球服務團隊,為醫療器械(含IVD等)企業提供產品注冊認證、大動物試驗、臨床試驗及孵化轉化等全球市場合規準入的全流程服務。
廣州:廣州市荔灣區花地大道中228號佛山:佛山市南海區北湖二路雅致廣場珠海:珠海市橫琴新區橫琴濠江路8號蘇州:蘇州市蘇州工業園區勝浦街道興浦路200號合肥:合肥市經濟技術開發區宿松路4090號
138 2641 2791
constant@constantmed.com
熱線電話
上班時間
周一到周五
公司電話










 客服1
客服1